
Copper Proteins for BioFuel Cell Applications
Recently there has been a renaissance in biological science and engineering. Powerful new technologies have emerged whose development cross the classic borders between biology, chemistry and engineering disciplines. A number of practical applications have resulted, including: sensors, drug dosing and making, combinatorial diagnostics and artificial organs. Using such devices in vivo is highly desired, and this is motivating the development of a means for generating electrical power that is compatible with biological environments. For in vivo applications, providing electrical power by using a fuel cell is especially attractive, because a fuel cell could indefinitely supply power to an implanted device by scavenging reactants from the sera in living organisms. Attractive as bio fuel cells may be, serious challenges remain. One challenge is finding catalysts to activate electrode reactions for power generation from chemical species found in sera, while avoiding interference from other species and maintaining stability in complex physiological fluids. Perhaps, the most important of these species is O2, because it is usually available, and is apt to be required, for the cathode in a bio fuel cell.
Iron-based hemoglobin, the heme center, itself, and heme-like molecules are often considered the most relevant catalytic centers for promoting oxygen chemistry in biological systems. The intention of this web page is to highlight another class of enzymes as catalytically active for oxygen chemistry as hemoglobin, namely the copper proteins, such as, the laccases, oxidases, tyrosinase and hemocyanin. These Cu enzymes and their synthetic analogues have promise and unique advantages for making bio fuel cell cathodes.
A structural unit often found in copper proteins that catalyze reactions of molecular oxygen is an antiferromagnetically coupled, two-copper-ion fragment. This two copper-ion fragment has been associated with the active sites of enzymes when they perform many remarkable phenomena, such as: 1) the reversible, binding of molecular oxygen, O2, during oxygen transport (e.g., by the enzyme called hemocyanin), 2) the catalysis of the electron transfers which lead to the oxidation of small molecules and the reduction of O2 to H2O (e.g., by the blue oxidases, such as, ascorbate oxidase, ceruloplasmin and the laccases), and 3) the catalytic insertion of an oxygen atom, O, into organic molecules, in which the source of O is O2 (e.g., by tyrosinase). Simple copper ions in aqueous solution are not nearly as active as catalysts for these reactions as are the copper proteins. Proposed active site structures and how they interact with O2, are shown below.
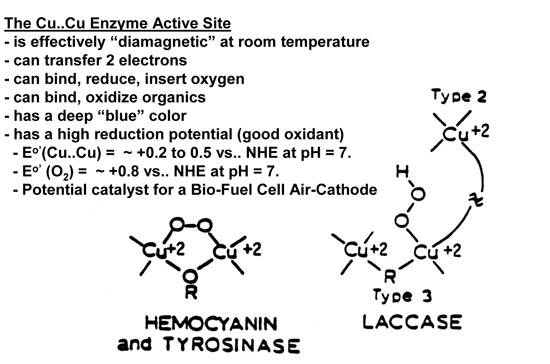
Hemocyanin And Tyrosinase
Hemocyanin and tyrosinase cycle between the cupric, Cu(II), and cuprous, Cu(I), oxidation states during biological functions. The binuclear cupric ions are so strongly antiferromagnetically coupled, that this Cu(II)-Cu(II) site in these proteins is effectively diamagnetic under anaerobic conditions at room temperature. Figure below summarizes the electronic and magnetic qualities of such a Cu(II)-Cu(II) site:
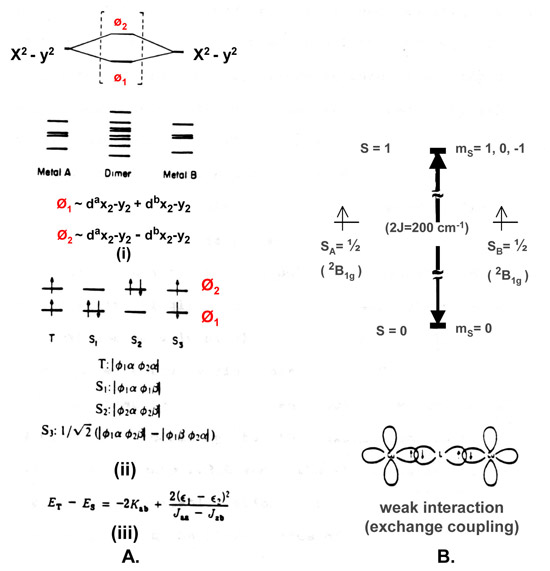
The molecular orbitals of Cu dimmer (Figure A above) are the in and out of phase combinations of the primarily d-atomic orbitals from the two monomeric copper ion fragments which make up the dimer. The 8 lowest energy dimer orbitals are filled by 16 of the 18 electrons available in the Cu(II)-Cu(II) oxidation state. Only the state corresponding to the T, S1 and S2 configurations need to be considered, because only these can possibly have appreciable populations up to 300K. The energy difference between the triplet state (T) and the singlet state (S), ET - ES, is equal to – 2 Kab + 2 [ ( є1 - є 2) 2 ]/ [Jaa – Jab ]. Kab, the kinetic exchange energy between Cu(II)a and Cu(II)b, is virtually constant and equal to ~50 cm-l. Jaa and Jab are the coulombic end exchange integrals, respectively, and their difference Jaa-Jab is also virtually constant and equal to ~ 40,000 cm-l. ET -ES is only strongly dependent on the square of the difference of the energy, є1 , of molecular orbital phi1 (Ø1, refer to Figure 2.a.(i) ) and the energy, є 2 , of molecular orbital phi2 (Ø2). The occupancy for the Boltzmann distribution of the triplet and singlet states at room temperature, 300K, is almost exclusively singlet (i.e., the Cu(II)-Cu(II) center is effectively diamagnetic) when ET -ES is ~ 700 cm-1. Figure B shows the stabilization of S = 0 state and the destabilization of the S = 1 states of the dimer relative to the S = 1/2 states of the two monomeric fragments for an antiferromagnetic interaction.
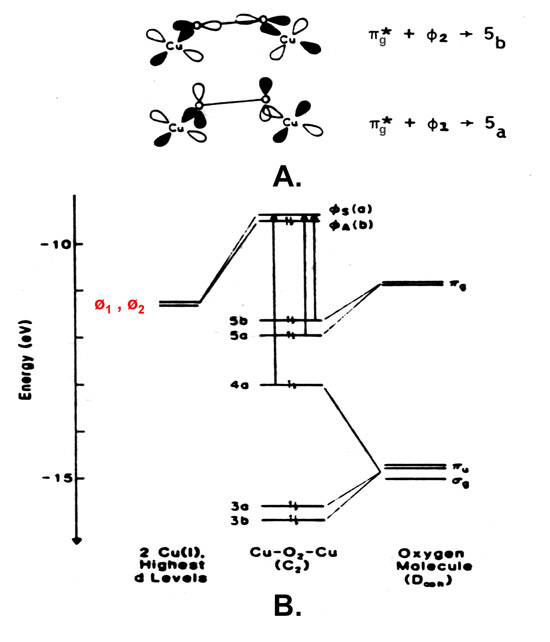
Anaerobic samples of hemocyanin or tyrosinase in the oxidized state, Cu(II)-Cu(II), do not show the 330 nm absorption band characteristically associated with proteins with a Type 3 copper center (Lena, make a link to the section beow) under similar conditions. The Cu(I)-Cu(I) fragment in the reduced form of hemocyanin or tyrosinase does however react reversibly with molecular oxygen, O2, to readily form a peroxide-bridged binuclear cupric fragment, which exhibits two characteristic new charge transfer transitions, one at ~350 and the other at ~570 nm. Figure below shows the pi (π) antibonding orbital of O2 overlapping the cuprous dimer orbitals in hemocyanin or tyrosinose and the subsequent molecular orbital diagram:
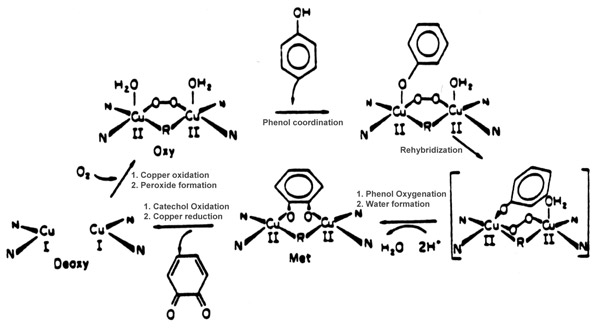
Although the entire biological function of hemocyanin is limited to reversibly binding O2, tyrosinase can also function as a mono-oxygenase as well as a two electron oxidase. When the tyrosinase Cu(I)-Cu(I) site binds O2 to make [Cu(II)-O-O-Cu(II)]+2, this peroxide bridged cupric fragment is a source of activated oxygen. The substrate, phenol, formally receives an oxygen atom from the [Cu(II)-O-O-Cu(II)]+2 fragment (e.g., a bound phenolate is converted to catecholate). The oxene depleted fragment, [Cu(II)-O-2...Cu(II)]+2, accepts two protons and liberates water. The water depleted cupric fragment which remains, [Cu(II)...Cu(II)]+2, accepts two electrons from the substrate (transforming catechole to quinone). This sequence of events regenerates the Cu(I)...Cu(I) fragment in the active site, which is then ready to undergo another catalytic cycle. This proposed scheme for tyrosinase activity is summarized in Figure below:
Type 3 Centers
Copper ion active sites in enzymes and proteins have been classified as belonging to the categories called the Type 1, Type 2 or Type 3 copper centers. In the native enzymes these three centers can be found either together or individually. Both the Type 1 and Type 2 centers denote sites containing only one copper ion. The Type 3 center was initially described as a copper cation site which (i) can accept two electrons, with the reduction potential for site being ~ 400 to 800mV versus the Normal Hydrogen Electrode at pH of ~ 7, (ii) is effectively diamagnetic at room temperature and shows no electron paramagnetic resonance signal at 77 K while the site is in its oxidized state, and (iii) absorbs 330 nm ultraviolet radiation with an absorption coefficient of ~ 2,000 to 4,000 M-1 cm-1. Sensitive, variable temperature, magnetic susceptibility measurements confirmed that the oxidized state of this site has two Cu(II) ions which are antiferromagnetically coupled so strongly that this active site should be effectively diamagnetic at room temperature.
Enzymes with a Type 3 center (e.g., the blue oxidases: ascorbate oxidase, ceruloplasmin and the laccases) catalyze the oxidation of substrates with molecular oxygen, O2, being the ultimate oxidant. As the substrate is oxidized, the Cu(II)-Cu(II) fragment can accept two electrons to form a Cu(I)-Cu(I) fragment. This Cu(I)-Cu(I) fragment in the fully reduced enzyme is reoxidized by O2 thereby forming H2O. This is summarized below:
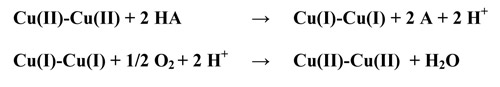
Molecular oxygen, O2, is incapable of reoxidizing the Cu(I)-Cu(I) fragment found in the Type 3 center if a Type 2 center is not present. Therefore the Type 2 center is associated with the working Type 3 site.
It is remarkable, that the apparently simpler binuclear copper ion active site in tyrosinase, can accommodate reversible oxygen binding, oxygenation end oxidation catalysis, while only oxidation catalysis is associated with the apparently much more complicated multicopper active site structures found in the blue oxidases.
Acknowledgement

The content of this web page is created by Dominic Gevasio. Photo of Natalie Hanson with a protein structure image taken from the web site of Alexandre de Breven.
More on proteins
- Copper Proteins - Wikipedia Article
- Copper Proteins - Classification
- The Copper Proteins
- Enzyme Design
- Microbial Fuel Cell - Wikipedia Article
- Hemoglobin and Myoglobin