
Enzyme Design
1. What are enzymes and who cares?
 Enzymes are proteins that catalyze various reactions in organisms. For example, when your liver metabolizes drugs, caffeine, or alcohol, it uses an enzyme called P450 to speed up the process. The author of this page is surely thinking about that over the bottle of Chianti (below). Enzymes are responsible for transporting oxygen down your blood-stream, digestion, and production of hormones (such as cholesterol, adrenaline,
estrogen and testosterone). Plants use enzymes to split nitrogen and make it bioavailable, and to convert CO2 to O2 during photosynthesis. Enzymes are also the survival weapons of bacteria, for example the one shown at right belongs to Mycobacterium Tuberculosis. Life on Earth is indeed "engined" by enzymes.
Enzymes are proteins that catalyze various reactions in organisms. For example, when your liver metabolizes drugs, caffeine, or alcohol, it uses an enzyme called P450 to speed up the process. The author of this page is surely thinking about that over the bottle of Chianti (below). Enzymes are responsible for transporting oxygen down your blood-stream, digestion, and production of hormones (such as cholesterol, adrenaline,
estrogen and testosterone). Plants use enzymes to split nitrogen and make it bioavailable, and to convert CO2 to O2 during photosynthesis. Enzymes are also the survival weapons of bacteria, for example the one shown at right belongs to Mycobacterium Tuberculosis. Life on Earth is indeed "engined" by enzymes.
Not only are enzymes diverse and very specific in functionality, they are also highly efficient catalysts operating at mild conditions. Synthetic catalysts are almost never comparably efficient. It is therefore very tempting to use some of the Nature's profound catalytic strategies in artificial catalysts. Wouldn't it be nice to be able to come up with new proteins capable of speeding up any reaction we want?
2. How do enzymes work?
So how do enzymes catalyze chemical reactions? First, let us state this: chemical reactions are much like hiking. Yes, indeed. Take a look:
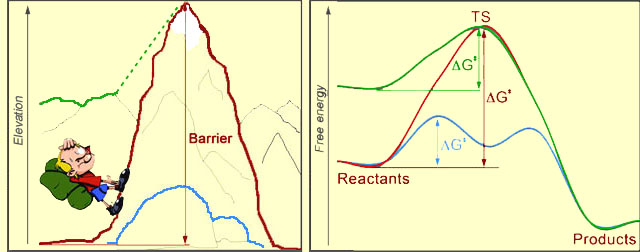
At left, you see a hiker, who's about to climb the mountain. Sure seems like a lot of work! In the picture at right, the red curve represents a chemical reaction, transforming "Reactants" to "Products". The reaction would require an input of energy, called the activation barrier (ΔG‡ - Delta G double-dagger). The magnitude of the barrier is the difference between the levels of the reactants and uppermost point on the red curve, which is called the Transition State (TS) of the reaction. The bigger the barrier is, the harder it is for the process to proceed. After TS, rolling down-hill is easy, both for the hiker and for the chemical system.
 The reaction rate is an exponential function of the negative activation barrier. Therefore, if we want to speed up a reaction (catalyze it), we need to lower the barrier. This can be done either by lowering the energy of TS (blue curve), or by increasing the energy of the starting point (green curve). Just so, the hiker could choose a different path to the other side of the mountain: he could walk over the blue boulders, or he could start his journey from the nearby green mountain, which is already tall.
The reaction rate is an exponential function of the negative activation barrier. Therefore, if we want to speed up a reaction (catalyze it), we need to lower the barrier. This can be done either by lowering the energy of TS (blue curve), or by increasing the energy of the starting point (green curve). Just so, the hiker could choose a different path to the other side of the mountain: he could walk over the blue boulders, or he could start his journey from the nearby green mountain, which is already tall.
We just revealed the two major strategies that enzymes use to catalyze chemical reactions: Enzymes either tweak the transition state so as to lower it's energy (stabilize it), or they destabilize the reactants. The barrier thus gets reduced, and the rate of the reaction goes up.
How exactly do enzymes do it? They wisely use their structure!
Any enzyme has a binding pocket, or binding site of a specific structure, such that the transition state of the catalyzed reaction sits there comfortably. The size and shape of the binding pocket are just right to accommodate a given transition state. And the amino acids that point into the binding pocket favorably interact with the transition state. This way the TS is pulled down in free energy, thereby reducing the barrier to the chemical reaction.
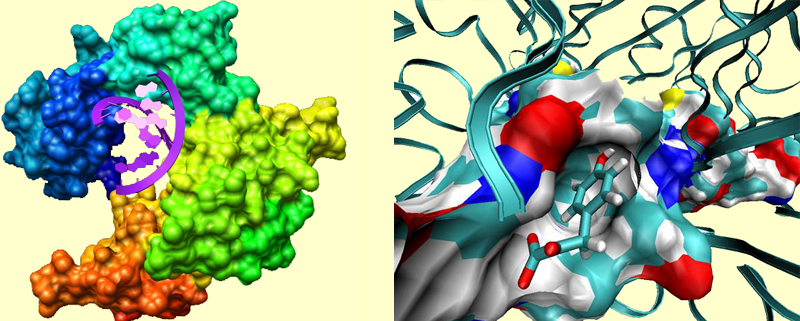
For example, in the picture at left, an enzyme called DNA-polymerase binds a chunk of DNA. Notice how the protein is shaped in order to accommodate the DNA double helix. In the picture at right, you see a binding site of a protein called MIFF, which is a pharmaceutical target for cancer and inflammatory diseases. A drug molecule is bound to the active site. At this close view, you can notice several profound interactions between the protein and the substrate. The blue regions in the surface of the binding pockets represent more electrophilic parts (such as protonated Lys), and the red regions represent more electrophobic parts (such as deprotonated Asp). The functional groups such as carbonyls and hydroxyls often point toward the bluer regions, amino groups point toward red regions, and more indifferently, non-polar parts of the substrate reside in nonpolar regions of the pocket.
Not only does the structure matter, but also the nature of the functional groups present in the site (they will do the catalysis). For example, look at the binding site of metallo-β-lactamase (below). This protein is present in bacteria, and it abolishes the activity of β-lactam antibiotics helping bacteria to survive. This enzyme hydrolyzes the β-lactam unit in the antibiotics molecule. The active site is fairly open and exposed to the solvent, though the protein loop located above the active site is thought to loosely close over the bound substrate. The inset shows the site with the bound β-lactam.
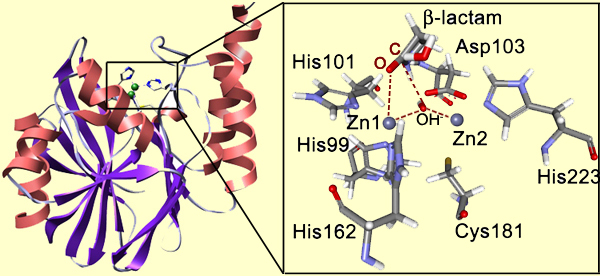
Notice that Zn1 coordinates the carbonyl oxygen atom. This provides positioning and polarization of the substrate: the carbonyl carbon is exposed for the nucleophilic attack and its positive charge is increased. In turn, the bridging OH- group is also positioned and polarized by the Zn2+ cations so that it is a very strong nucleophile ready to perform the attack on the carboxyl carbon. So the attack happens very easily, whereas in solution it is the rate-determining step of the β-lactam hydrolysis. The reduction of the barrier to the nucleophilic attack is the essence of the catalysis. Here is the full mechanism of the enzyme catalyzed reaction:
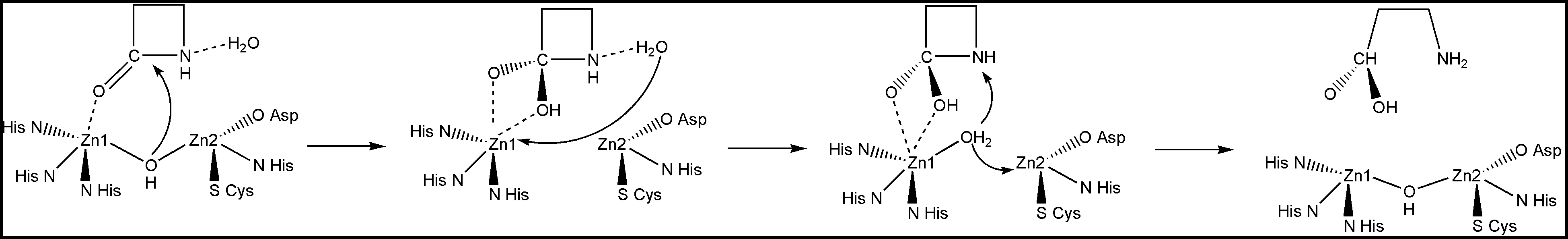
These are just some of the Nature's strategies. There are many more of them in Nature. We can use them, or, of course, we can come up with our own new strategies, and implement them in the design of artificial enzymes.
3. So, how to design an enzyme?
Putting it all together: in order to design a new enzyme:
- Design a protein that binds and stabilizes the transition state of a reaction of interest.
- Introduce chemical functionality that will run the reaction in the protein.
- Make sure the product of the reaction also gets well-bound in the site, but not too strongly so as to prevent product inhibition.
Complications:
- Proteins are huge flexible objects with very many degrees of freedom. The task of enzyme design is extremely multi-variable.
- You still need some chemical intuition in order to incorporate the right kind of chemistry in the artificial enzyme.
Encouragement:
- Enzyme design is difficult but proven to be possible.
Enzyme design is like drug design turned inside-out. When you design a drug, you come up with a molecule that would bind strongly to an active site of a protein. In enzyme design, you come up with a protein that would bind strongly a molecule of choice (TS).
Enzyme design is also mechanistic enzymology turned inside-out. Instead of identifying the mechanism of a catalyzed reaction, you fix the mechanism (by choosing TS), and then build a protein that could accomplish it.
Recently an efficient protocol has been developed for the design of new enzymes. Not surprisingly, it is called the inside-out protocol.
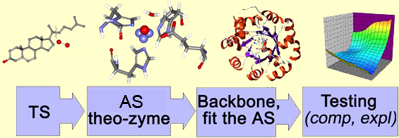
We aim at the transition state stabilization. The protocol thus begins with a tentative structure of TS found using ab initio techniques. We then use our chemical intuition and place various amino acids around the found transition state, such that the interactions are beneficial and amino acids can do the desired chemistry. Finally, a robust natural protein fold is selected, such that it could accommodate our transition state, and catalytic amino acids in the desired orientations. The native amino acids in the cavity of the protein will be mutated so as to turn a protein into an artificial enzyme. The enzyme would further be tested for the catalytic activity, both in silico, and in vitro.
4. Let us design an enzyme
Here is how the inside-out protocol can be used, step by step, to design a new enzyme. Let us take Kemp elimination as a reaction of interest:
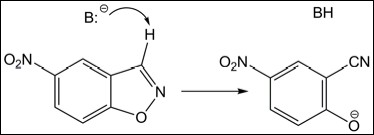
A general base, B:-, abstracts the proton from the substrate, the N-O bond opens, and, finally, O- gets protonated completing the reaction. The first step (shown in the Scheme above) is slow and rate-determining. Therefore, it should be the focus of catalysis.
In solution, this reaction can be easily catalyzed by strong acids or bases. However, in the protein environment, this is not possible; all we have is the 20 amino acids. How would we build the protein catalyst? Here is one strategy:
Let us use deprotonated glutamic acids, Glu, as the general base. This Glu should sit in the binding site awaiting the arrival of the substrate. As we mentioned, we need to stabilize the transition state of the rate-limiting step of the reaction. The TS consists of a molecule of the substrate with partially open N-O bond and H+ being partially transferred to the base (Glu) (Figure 1):
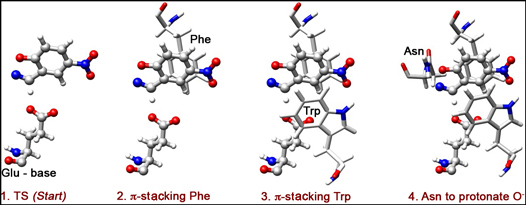
Now, how can we "comfort" the TS, i.e. species shown in Figure 1? How can we lower its energy? First of all, we need to keep the H-bond contact between the substrate and the base short, so as to promote an efficient H-transfer and thus lower the energy of the TS. Additionally, the O atom of the substrate starts accumulating negative charge as the N-O bond opens. Therefore, electrophilic residues interacting with this charge could help TS-stabilization.
One possible strategy: we can, introduce aromatic residues that would interact with the substrate in a π-stacking mode. Here, we opt to put phenylalanine (Phe) on the back of the substrate (Figure 2), and triptophan (Trp) on the front (Figure 3). These two residues will stabilize the negative charge developing on O via π-delocalization, and also form a slot for the substrate to dive into, toward its contact with the catalytic base (Glu).
An additional stabilization of O- can be done through H-bonding. Ultimately, we need a proton-donor in the region of O-, so as to complete the reaction by protonation. Here, we choose Asn as a H-bond donor (Figure 4).
This is the theo-zyme: theoretically designed active site of an enzyme. Now, we need to place this set of catalytic residues in the desired orientations into a protein, and create such an environment of the binding site as to preserve the protonation states of the important residues. We think that having a generally hydrophobic binding site is good for the catalysis. So we want our catalytic residues to be surrounded by a matrix of well-packed hydrophobic residues, such as Lue, Ile, Val, Gly, Phe, and Ala. Fortunately, now the task of residue-selection and re-packing is automated. In the Figure below we show the result:
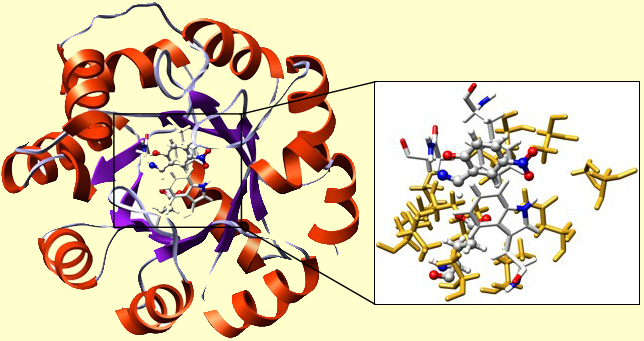
A TIM-barrel protein fold was used. This is one of the most robust folds, capable of withstanding multiple mutations. The cavity of the TIM was re-built. In the inset, you may find our TS in a H-bond contact with Glu. There you can also see Phe, Trp, and Asn, just as we placed them. The surrounding 'yellow stuff' are well-packed residues, forming the hydrophobic microenvironment of the active site. Their precise chemical nature is insignificant for the functioning of the present enzyme.
The design is done. Now, we need to test this new enzyme, first in silico and then in vitro. First, we run the catalyzed reaction computationally. Using the Free Energy Perturbation methodology, and QM/MM Monte Carlo, we can build the free energy surface for the reaction. In the picture at left, you see the QM/MM MC setup: a protein in a water droplet (computationally solvated). The picture at right shows the free energy map for the reaction, where the bluer parts are lower in free energy, and redder are higher in free energy.
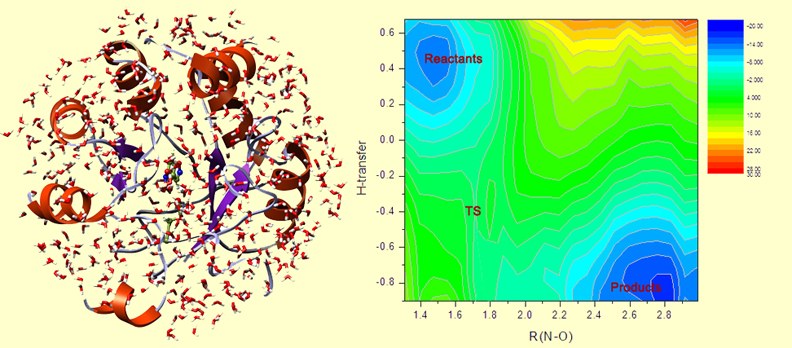
On this map, the reactants are in the upper-left corner. The N-O bond opens to the right, and the H-transfer downward. And the lower-right corner corresponds to the products. This map slightly asymmetric with respect to the diagonal in the map, which indicates that H-transfer happens a bit earlier and triggers the subsequent opening of the N-O bond.
The difference in free energy between TS and the reactants is the activation barrier. In this case, it is 13.5 kcal/mol. We can compare it to the barrier to the uncatalyzed Kemp elimination in water, which is ~20 kcal/mol. 13.5 is less than 20, therefore, we detect a catalytic effect of the protein! The reaction proceeds faster in this artificial enzyme than in water.
Experimental testing of this design was also done. The protein was expressed in E.Coli, purified, and tested to catalytic activity in vitro. The catalytic effect was confirmed. kcat / kuncat was ~102. This is not a huge catalytic effect, compare to some of the natural enzymes. However, it is a good start, and this success promises a big future to the field of enzyme design.
5. Where to read more on enzyme design?
There are many other successful designs out there. For example, artificial retro-aldolases and triose phosphate isomerases were reported recently. Visit the pages of the laboratories working on enzyme design for more information:
- David Baker, The University of Washington
- Steve Mayo, Caltech
- Donald Hilvert, Swiss Federal Institute of Technology, Zurich
- Ken Houk, UCLA
- William Jorgensen, Yale University
Acknowledgement
This page was created by Anastassia Alexandrova, Postdoctoral Associate in the laboratory of Professor William Jorgensen at Yale University.