
Aromaticity
“Aromaticity” is another ill-defined term used in chemistry, just like “chemical bond” itself. At some point chemists (or more precisely, physicist Michael Faraday) synthesized a compound that they could not describe on the basis of the contemporary knowledge of chemical structure. The compound called “benzene” had very low ratio between hydrogen and carbon atoms - it was highly unsaturated. It also had a strong aroma, sometimes recognized as similar to the smell of old gloves. Very soon the entire class of substances was singled out: they all were highly unsaturated and fragrant, derived from essential oils, resins etc. Due to the lack of the actual information about the chemical structure and the origin of chemical properties, these substances were called “aromatic” – the easily recognizable common feature was used for pure labeling purposes.
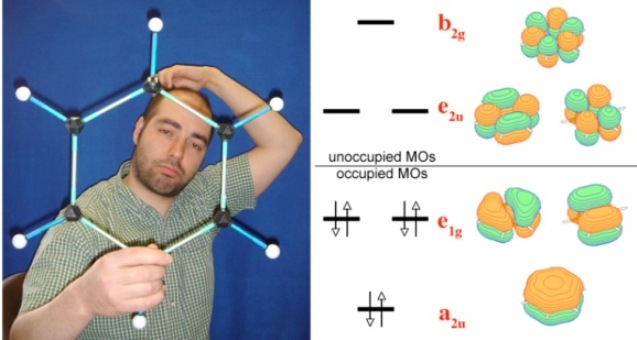
Model of the benzene molecule and diagram of the valence ?-orbitals of benzene responsible for the delocalized (aromatic) bonding.
The concept of antiaromaticity originally was accepted and established in organic chemistry. As of year 2001, it was extended to the realm of all-metal clusters and later main-group element and transition metal clusters. It is relevant to the chemistry of nanosystems such as nanotubes, nanospheres, nanowheels, nanosprings etc.
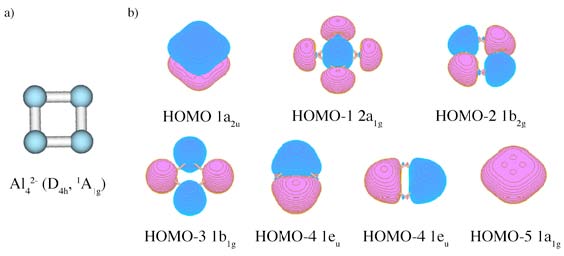
a) structure of an isolated Al42- unit; it is a part of NaAl4– cluster observed in molecular beam;
b) Canonical Molecular Orbitals of Al42–(D4h, 1A1g) structure; HOMO1a2u is π–aromatic orbital, HOMO–1 2a1g is radial σ–aromatic orbital and HOMO–2 1b2g is
tangential σ–aromatic orbital.
The development of methods of structural analysis together with evolution of the theoretical insights into the world at atomic and molecular scale led to the elucidation of the peculiarities of the electronic structure of aromatic compounds. In chemistry the structure of molecules is represented using the concept of chemical bonds, where a pair of electrons is associated with two atoms (see Lewis Model of Bonding). It turns out that in aromatic compounds it is impossible to make such association. Certain amount of valence electrons has to be associated with all the atoms in the molecule. In other words, these electrons cannot be localized and the structure of the molecule cannot be uniquely represented using only pair-wise interactions between atoms – 2-center 2-electron (2c-2e) bonds. So, from the bonding point of view aromaticity is equivalent to the delocalization of certain amount of valence electrons.
There are two approaches to the description of the electronic structure of aromatic systems. First one is based on the theory of resonances. If there is no unique representation involving only 2c-2e bonds between atoms, one might think of several representations being in “resonance” or “mixing” with each other. It is important to keep in mind though, that there are no real resonance structures rapidly transforming into each other. “Mixing” occurs between several non-unique representations of a single real system. From the theoretical point of view, these resonance representations and their relevance can be obtained through computations based on valence bond theory.
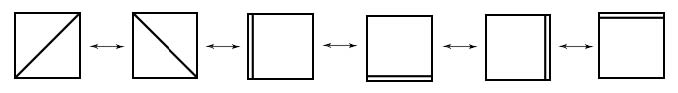
Resonance (Lewis) structures contributing to the description of aromatic Al42 –(according to Havenith, R. W. A., van Lenthe, J. H. Chem. Phys. Lett. 2004, 385, 198).
The second approach considers molecular orbitals (MOs), or eigenstates of some energy related operator (see Molecular Orbital Theory and Self-Consistent Field Approximation). Molecular orbital characterizes spatial distribution of a pair of electrons with anti-parallel spins. So, by their very nature, MOs are delocalized over the entire molecule. It turns out though that certain sets of MOs can be transformed into lone-pairs (LPs) and 2c-2e bonds (see Natural Bonding Orbital Analysis). In aromatic systems there are also sets of MOs that cannot be transformed in this manner and should remain completely delocalized.
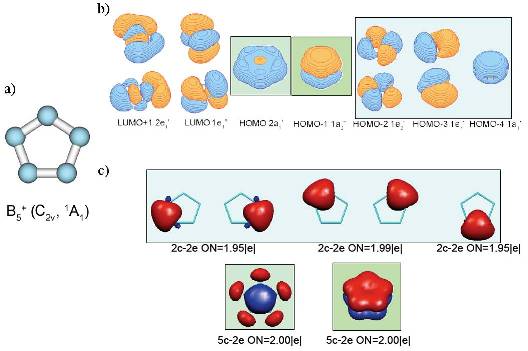
a) geometry of the most stable isomer of B5+. Though it is supposed to have five-fold rotational axis and possess D5h symmetry, the second-order Jahn-Teller effect leads to the structural distortion resulting in C2v symmetry of the global minimum
structure; b) Canonical Molecular Orbitals of B5+ (D5h, 1A1‚) structure; higher symmetry is chosen to preserve degeneracy of CMOs; c) localized description of the electronic structure of B5
+(C2v,1A1) cluster obtained by Adaptive Natural Density Partitioning;
color-coding shows relations between localized multi-centered bonds and CMOs from part (b); it is clearly recognizable, that 5c-2e bonds of σ– and π–nature make the system doubly aromatic.
The delocalization of bonding has one important implication. Perfect delocalization requires atoms (or fragments) of a chemical system to be equivalent in geometric sense, i.e. meet certain symmetry requirements (see Symmetry). As the result, aromatic systems are very often highly symmetric and, in its turn, high symmetry can be a hint that the system is aromatic. High symmetry usually (but not always!) means presence of a multifold rotation axis in case of planar systems or polyhedral cage-like structure of Td, Oh, and Ih symmetry in case of 3D geometries (spherical aromaticity).
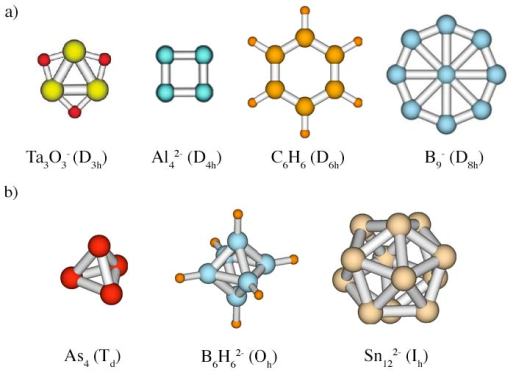
Examples of a) planar (2D) and b) spherical (3D) highly symmetric aromatic systems.
Also, as long as electronic structure is considered, the perfect delocalization and high symmetry can be preserved only if MOs are occupied according to certain rules (see Hückel Aromaticity and Mobius aromaticity). These rules are based on the requirement that all degenerate (having the same energy) MOs involved into aromatic bonding should be completely occupied. If this requirement is not met, the first-order Jahn-Teller effect leads to the structural distortions and formation of islands of aromaticity in the system, which is now globally anti-aromatic.
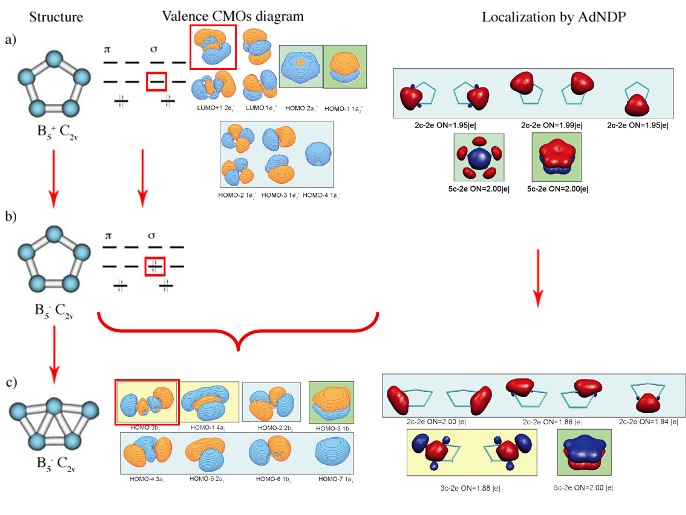
First-order Jahn-Teller distortion of a) doubly (σ– and π–) aromatic B5+ (C2v) cluster into b) B5- C2v system with partially filled degenerate orbital upon addition of an extra electron pair and further into c) σ–antiaromatic π–aromatic B5- C2v cluster. Canonical Molecular Orbitals at (a) and (b) correspond to D5h symmetry so that the degeneracy of orbitals is preserved for illustrative purposes. Structure (a) B5+ initially has C2v symmetry due to the second–order Jahn–Teller distortion. Evolution of the bonding pattern and formation of islands of σ–aromaticity in (c) is revealed using AdNDP
analysis.
Based on the nature of the overlap in the MOs forming aromatic system in planar molecules, we distinguish between σ– aromaticity, π–aromaticity, δ–aromaticity (these three types were actually spotted in real systems) and φ–aromaticity (predicted but not encountered so far).
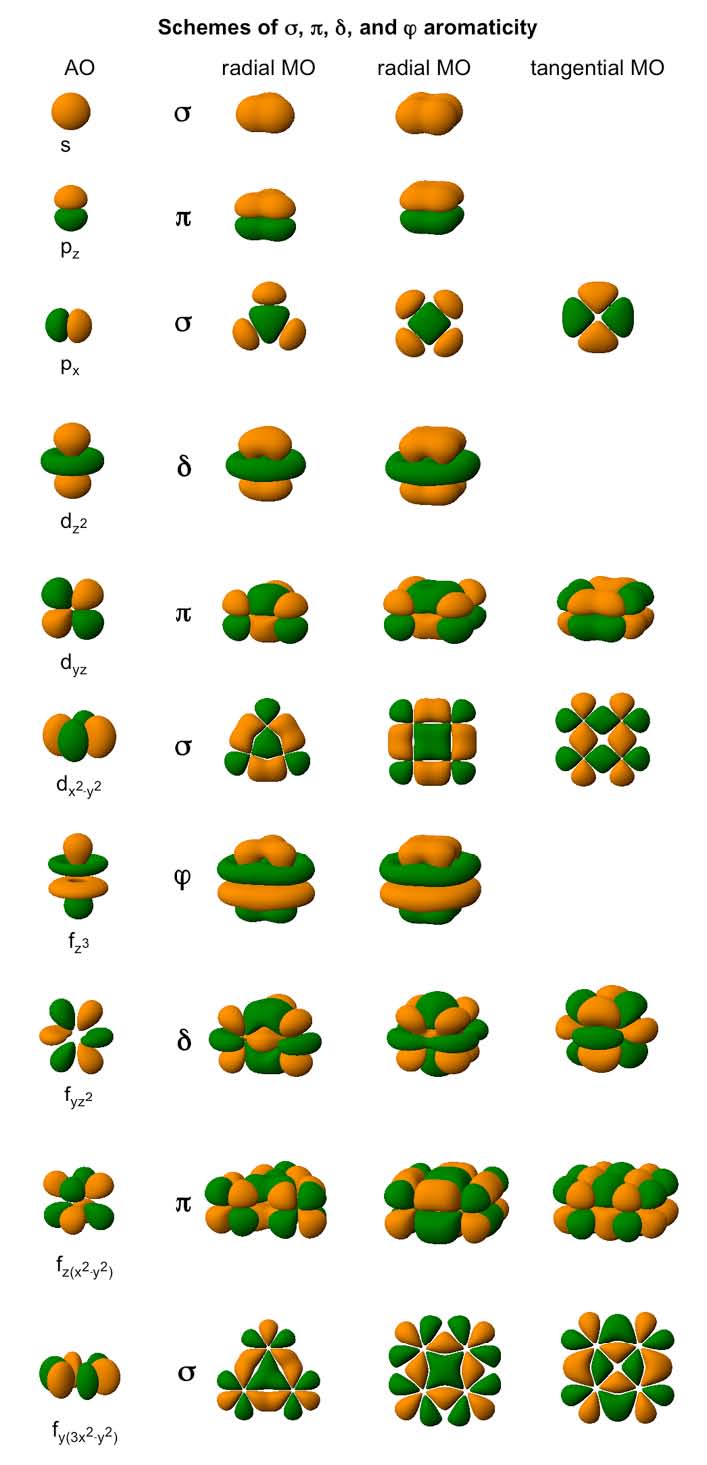
Various types of completely bonding (aromatic) MOs in a generic three–atomic system.
Can certain molecule or cluster possess several types of aromaticity? The answer is yes. While aromaticity/antiaromaticity in organic systems is usually of π–type, the key feature of bonding in subnano- and nano-clusters is multifold nature of aromaticity. Think of all possible combinations of σ–and δ– aromatic/antiaromatic bonding. They all can be found in planar or quasi-planar clusters of main group elements! For example, B5+ is σ– and π– aromatic, B5- is σ–antiaromatic and ?-aromatic, and B62- is σ– and π– antiaromatic and so on. Now, throw in δ– aromaticity/antiaromaticity, which can be present in transitional metal clusters. The number of possible combinations will be even bigger! In spherical clusters where σ/π/δ separation is not possible there are still different types of aromaticity: radial and tangential, according to the location of the overlap regions of MOs responsible for the delocalized bonding.
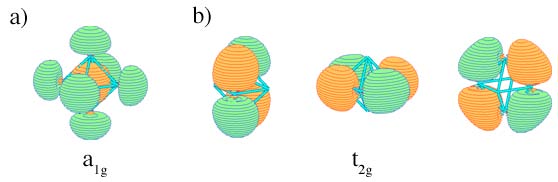
Examples of MOs with a) radial overlap and b) tangential overlap in a generic system of Oh symmetry.
In old days a substance would be assigned as aromatic if it had an aroma. Many of the substances considered as aromatic by contemporary chemists are not fragrant at all. Aromatic nature of chemical bonding can be established by analysis of electronic structure. To identify certain cluster as aromatic/antiaromatic it is necessary to outline the sets of the MOs that can’t be transformed into the classical Lewis bonding objects, such as lone pairs and 2c-2e bonds. These orbitals are compared with prototypical aromatic systems on the basis of overlap type and separated into subsets, corresponding to different types of aromatic bonding (e.g. σ, π, δ). Than aromaticity/antiaromaticity is established on the basis of counting rules, specific for the delocalized bonding of each type. In this way it was possible to develop comprehensive model of chemical bonding for entire family of planar and quasi-planar boron clusters.
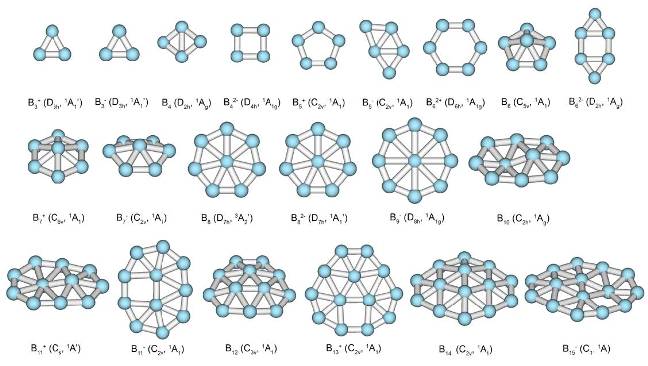
Family of planar and quasi-planar boron clusters (according to Zubarev, D. Yu.,Boldyrev, A. I. J. Comput. Chem. 2007, 28, 251.)
Are there any observable properties that can be used for distinguishing aromatic systems from other (non-aromatic)? The additional stabilization energy (compared to the virtual structures with localized bonding) is a good measure of aromaticity called resonance energy. Delocalized bonding is energetically more efficient than localized and it is more important for the stability of a molecule, so aromatic molecules should be inert in the reactions involving the delocalized bonds. Delocalization of the electrons over the entire molecule should lead to certain type of behavior in the presence of external magnetic field. Nuclear Independent Chemical Shift (NICS) indexes and induced ring currents rely on this phenomenon when trying to estimate degree of the aromaticity of the system. While useful for labeling purposes, these measures of aromaticity depend on many factors and peculiarities of electronic structure and very often contradict each other.
On the other hand, assignment of aromaticity on the basis of analysis of chemical bonding can easily establish connection between structure of a certain system and its chemical behavior, but this procedure does not rely on any observable features. Co-existence of various approaches to the definition of aromaticity (on the basis of electronic structure, resonance energy, magnetic properties, and chemical behavior) makes this topic ill-defined and somewhat controversial. Nevertheless, success of this concept in organic chemistry makes it very useful.

Acknowledgement
Alex Boldyrev (inspiration), Dmitry Zubarev (design) and Boris Averkiev (photo model)
Suggested reading
- 1. Minkin, V. L.; Glukhovtsev, M. N.; Simkin, B. Ya. Aromaticity and Antiaromaticity. Electronic and Structural Aspects; John Wiley & Sons, Inc.: New York, Chichester, Brisbane, Toronto, Singapore, 1994.
- 2. Bersuker, I. The Jahn-Teller Effect; Cambridge University Press, Cambridge, 2006.
- 3. Li, X.; Kuznetsov, A. E.; Zhang, H. F.; Boldyrev, A. I.; Wang, L. S. "Observation of all-metal aromatic molecules" Science, 2001, 291, 859.
- 4. Fowler, P. W.; Havenith, R. W. A.; Steiner, E. "Unconventional ring currents in an 'all-metal aromatic', Al-4(2-)" Chem. Phys. Lett. 2001, 342, 85.
- 5. Havenith, R. W. A., van Lenthe, J. H. "A valence bond study of the sigma and pi aromatic species Al-4(2-)" Chem. Phys. Lett. 2004, 385, 198.
- 6. Zhai, H. J.; Kuznetsov, A. E.; Boldyrev, A. I.; Wang, L. S. "Multiple Aromaticity and Antiaromaticity in Silicon Clusters" ChemPhysChem, 2004, 5, 1885.
- 7. Boldyrev, A. I.; Wang, L. S. "All-metal aromaticity and antiaromaticity" Chem. Rev. 2005, 106, 3716.
- 8. Alexandrova, A. N.; Boldyrev, A. I.; Zhai, H. J.; Wang, L. S. "All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry" Coord. Chem. Rev. 2006, 250, 2811.
- 9. Zubarev, D. Yu., Boldyrev, A. I. "Comprehensive analysis of chemical bonding in boron clusters" J. Comput. Chem. 2007, 28, 251.
- 10. Zubarev, D. Yu.; Averkiev, B. B.; Zhai, H. J.; Wang, L. S.; Boldyrev, A. I. "Aromaticity and antiaromaticity in transition-metal systems" Phys. Chem. Chem. Phys. 2008, 10, 257..